SynFlow: A workflow for synteny and chromosomal rearrangement
![]()
![]()
Table of Contents
- Overview
- Software requirements
- Installation
- 1. Install pixi
- 2. Clone the repository
- 3. Install dependencies
- Pixi usage
- SLURM execution
- Using snakemake.sh
- SLURM profile (slurm/config.yaml)
- Configuration
- Workflow config file (config.yaml)
- Workflow steps
- Output files
- Primary outputs
- Intermediate files (tmp/)
- Troubleshooting
- Citation
- License
Overview
A Snakemake workflow for synteny detection and chromosomal rearrangement analysis between two or more genome assemblies.
Software requirements
All dependencies are managed by pixi. No manual conda/pip setup is needed.
| Tool | Version | Purpose |
|---|---|---|
| Snakemake | 7.32.4 | Workflow management |
| SyRI | ≥1.7.1 | Synteny & rearrangement detection |
| minimap2 | ≥2.30 | Fast sequence alignment |
| MUMmer4 | ≥4.0.1 | Nucmer genome alignment |
| gffread | ≥0.12.7 | GFF/GTF processing |
| DIAMOND | ≥2.1.24 | Protein sequence alignment |
| MCScanX | ≥1.0.0 | Collinearity detection |
| Biopython | ≥1.83 | Sequence utilities |
| --- |
Installation
1. Install pixi
curl -fsSL https://pixi.sh/install.sh | bash
Restart your shell or run source ~/.bashrc (or ~/.zshrc) to make pixi available.
2. Clone the repository
git clone https://gitlab.cirad.fr/agap/cluster/snakemake/synflow.git
cd synflow
3. Install dependencies
pixi install
This reads pixi.toml and installs all tools into an isolated environment under .pixi/.
Pixi usage
Pixi manages the environment and provides shortcut commands. You do not need to activate any environment manually.
# Install or update the environment
pixi install --manifest-path pixi.toml
# Run snakemake through the pixi environment
pixi run snakemake --configfile config.yaml --cores 8
# Open an interactive shell inside the pixi environment
pixi shell
# Check installed tool versions
pixi run snakemake --version
pixi run minimap2 --version
SLURM execution
Using snakemake.sh
The snakemake.sh script is designed for HPC submission via SLURM. It handles:
- Automatic pixi installation if not present on the node
- Environment setup (
pixi install) before running - Snakemake dispatch with a set of predefined commands
# Submit to SLURM
sbatch snakemake.sh run
# Dry-run (check the workflow without executing)
sbatch snakemake.sh dry
# Generate DAG graph (requires graphviz dot)
sbatch snakemake.sh dag
# Unlock a locked working directory
sbatch snakemake.sh unlock
# Delete all outputs (with confirmation)
sbatch snakemake.sh clear
Available options:
| Option | Default | Description |
|--------|---------|-------------|
| -p PROFILE | slurm | Snakemake profile directory |
| -j NJOBS | 200 | Maximum number of SLURM jobs |
| -c NCORES | 700 | Maximum total cores |
| -r FILE | config.yaml | Workflow config file |
| -o FILE | images/dag.png | DAG output image |
Example:
sbatch snakemake.sh run -p slurm -j 50 -c 200 -r config.yaml
The script wraps snakemake so that all calls go through pixi run, ensuring the correct environment is used even on compute nodes that do not have snakemake in their PATH.
SLURM profile (slurm/config.yaml)
The SLURM profile controls job submission parameters:
cluster:
mkdir -p logs/ &&
sbatch
--partition={resources.partition}
--account={resources.account}
--cpus-per-task={threads}
--mem={resources.mem_mb}
--time={resources.time}
--job-name=smk-{rule}-{wildcards}
--output=logs/{rule}-%j.out
--error=logs/{rule}-%j.err
set-resources:
- minimap2:mem_mb=32384
- minimap2:time=2000
- nucmer:mem_mb=32384
- nucmer:time=2000
- syri_minimap2:mem_mb=18096
- syri_nucmer:mem_mb=18096
default-resources:
- mem_mb=4096
- partition=cpu-dedicated
- account=dedicated-cpu@cirad
- time=60
set-threads:
- minimap2=8
- nucmer=8
- syri_minimap2=8
- syri_nucmer=8
Configuration
Workflow config file (config.yaml)
Create a config.yaml file (or copy from test/) with the following structure:
# --- Input genomes (required) ---
input_genomes:
genome1: "path/to/genome1.fasta"
genome2: "path/to/genome2.fasta"
# Add more genomes as needed
# --- Alignment method ---
method: "nucmer" # "nucmer" (default) or "minimap2"
# --- Nucmer alignment parameters ---
min_length_match: 100 # Minimum match length (-l)
min_length_cluster: 500 # Minimum cluster length (-c)
min_distance_extension: 500 # Minimum distance for extension (-b)
min_alignment_identity: 90 # Minimum identity % for delta-filter (-i)
min_alignment_length: 5000 # Minimum alignment length for delta-filter (-l)
# --- Minimap2 parameters (used only if method: minimap2) ---
preset: "asm5" # Minimap2 preset (e.g. asm5, asm10, asm20)
bandwidth: 500 # Bandwidth for chaining (-r)
secondary: "no" # Report secondary alignments (yes/no)
# --- Optional: GFF annotations (required for MCScanX) ---
# Provide GFF files for all genomes when chromosome counts differ
input_gff:
genome1: "path/to/genome1.gff3"
genome2: "path/to/genome2.gff3"
Notes:
- At least 2 genomes are required. All pairwise combinations are processed.
- input_gff is optional but required when any two genomes have different chromosome counts (triggers MCScanX).
- method applies only to the SyRI pipeline (same chromosome count pairs).
Workflow steps
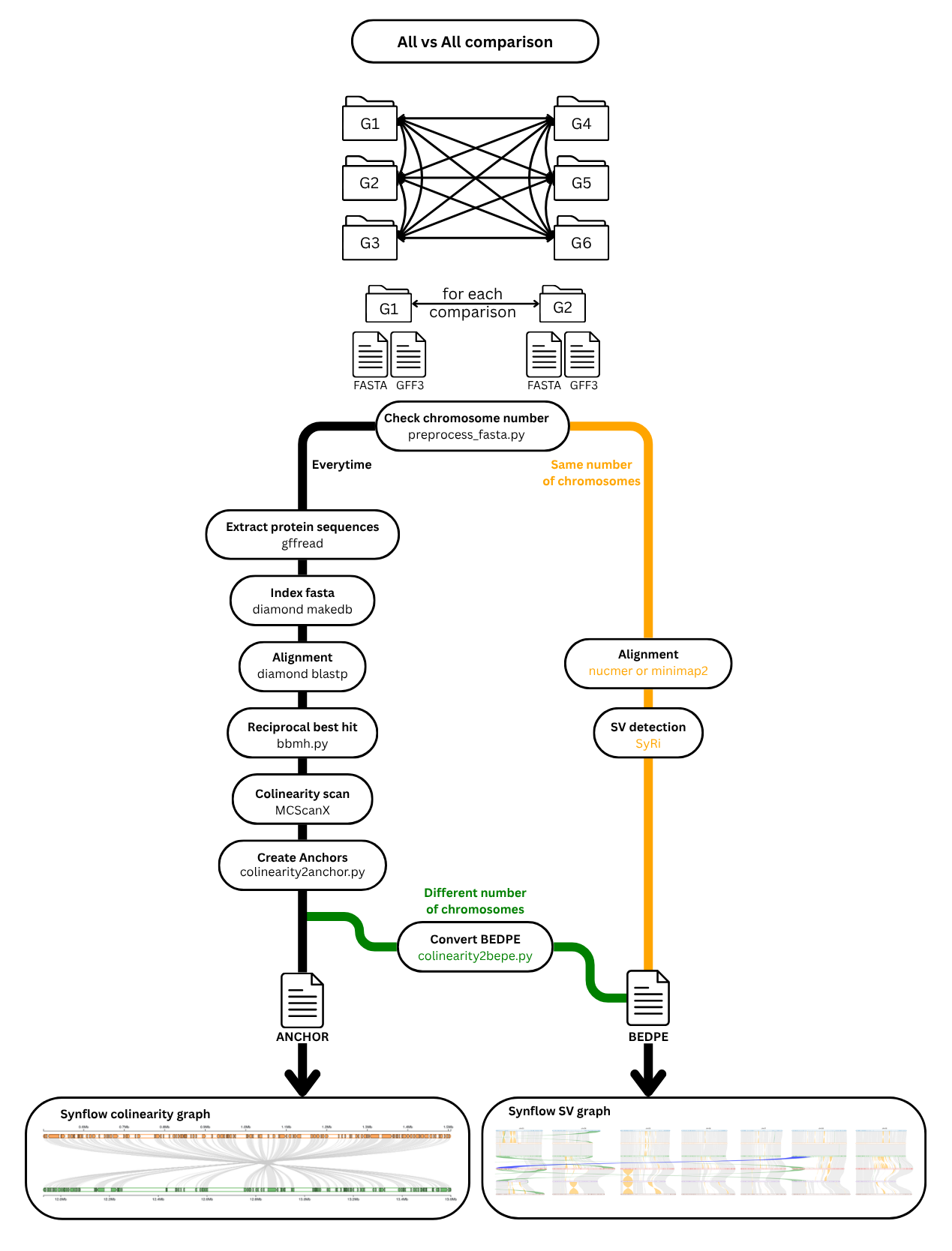
The workflow is organized as follows:
1. preprocess_fasta (checkpoint)
- Normalizes sequence IDs in each FASTA file (replaces special characters)
- Produces a JSON mapping of original → normalized IDs
- Counts the number of sequences (chromosomes/scaffolds) per genome
- Output:
tmp/{genome}_processed.fasta,tmp/{genome}_id_mapping.json,tmp/{genome}_chr_count.txt
2. gff2bed (optional)
- Converts GFF3 annotation files to BED format
- Applies the ID mapping from step 1 to normalize sequence names
- Output:
{genome}.bed,tmp/{genome}.gff3
3. gff2fasta (optional)
- Extracts protein sequences from the GFF3 annotations using
gffread - Output:
tmp/{genome}.prot
4a. nucmer (SyRI path)
- Aligns two genomes with
nucmer(MUMmer4) - Filters the delta file with
delta-filter(identity and length thresholds) - Generates a
.coordsfile for SyRI - Output:
tmp/mummer/{ref}_{qry}.filtered.delta,tmp/mummer/{ref}_{qry}.coords
4b. minimap2 (SyRI path, alternative)
- Aligns two genomes with
minimap2in SAM format - Output:
tmp/minimap2/{ref}_{qry}.sam
5. syri (SyRI path)
- Runs SyRI on the alignment output (nucmer or minimap2)
- Detects syntenic regions, inversions, translocations, duplications
- Restores original sequence IDs via
restore_ids.py - Output:
tmp/syri/{ref}_{qry}.out
6. diamond_prepdb + diamond_blastp (MCScanX path)
- Builds a DIAMOND protein database from the reference proteome
- Runs bidirectional BLASTP between both proteomes
- Output:
tmp/{genome}.prot.dmnd,tmp/diamond/{ref}_{qry}.out
7. bbmh4mcsanx (MCScanX path)
- Identifies best bidirectional hits (BBH) from the DIAMOND results
- Produces the
.homologyfile and a combined.gfffor MCScanX - Output:
tmp/{ref}_{qry}/{ref}_{qry}.homology,tmp/{ref}_{qry}/{ref}_{qry}.gff
8. mcscanx (MCScanX path)
-
Runs
MCScanX_hto detect collinear gene blocks -
Falls back to a looser parameter set if the output is below a size threshold
-
Output:
tmp/{ref}_{qry}/{ref}_{qry}.collinearity
9. collinearity2bedpe (MCScanX path)
- Converts the MCScanX collinearity file to BEDPE format (SyRI-compatible)
- Output:
tmp/mcscanx/{ref}_{qry}.out
10. collinearity2anchors (optional, MCScanX path)
- Extracts syntenic anchor gene pairs from the collinearity file
- Output:
{ref}_{qry}.anchors
11. finalize_bedpe
- Copies the appropriate result (SyRI or MCScanX) to the final output
- Output:
{ref}_{qry}.out
Output files
Tip: All primary output files (
.out,.bed,.anchors) can be directly loaded into the SynFlow web interface via the Upload Files section for interactive visualization.
Primary outputs
{reference}_{query}.out — Main synteny result (BEDPE format)
SyRI-style BEDPE output describing syntenic blocks and structural rearrangements between each genome pair.
- Columns 1–3: coordinates on the reference genome
- Columns 6–8: coordinates on the query genome
- Columns 9–12: structural annotation (SYN, INV, TRANS, DUP, etc.) For the full format specification, see: https://schneebergerlab.github.io/syri/fileformat.html
Example:
chr01 33618167 33780988 - - chr01 39357103 39440924 SYN2 - SYN BLOCK
chr01 33618167 33647271 - - chr01 39357103 39380755 Eg01_t020990 Macma4_01_g26690.1 SYN2 -
chr01 33725928 33733383 - - chr01 39391015 39394907 Eg01_t021030 Macma4_01_g26700.1 SYN2 -
{genome}.bed — Gene coordinates (if GFF provided)
BED file with gene positions for each genome, with normalized sequence IDs.
{reference}_{query}.anchors — Syntenic anchor pairs (if GFF provided, MCScanX path)
Tab-separated file listing syntenic gene pairs detected by MCScanX.
Intermediate files (tmp/)
| File | Description |
|---|---|
tmp/{genome}_processed.fasta |
Normalized FASTA |
tmp/{genome}_id_mapping.json |
ID mapping (original → normalized) |
tmp/{genome}_chr_count.txt |
Number of sequences |
tmp/{genome}.gff3 |
Filtered/normalized GFF3 |
tmp/{genome}.prot |
Extracted protein sequences |
tmp/mummer/{ref}_{qry}.coords |
Nucmer alignment coordinates |
tmp/mummer/{ref}_{qry}.filtered.delta |
Filtered nucmer delta |
tmp/minimap2/{ref}_{qry}.sam |
Minimap2 SAM alignment |
tmp/syri/{ref}_{qry}.out |
Raw SyRI output |
tmp/diamond/{ref}_{qry}.out |
DIAMOND BLASTP results |
tmp/{ref}_{qry}/{ref}_{qry}.collinearity |
MCScanX collinearity |
tmp/mcscanx/{ref}_{qry}.out |
MCScanX BEDPE output |
| --- |
Troubleshooting
- Memory errors: Increase
mem_mbfor alignment rules inslurm/config.yaml - Missing GFF files: Required for MCScanX when genomes have different chromosome counts
- Locked directory: Run
sbatch snakemake.sh unlockorpixi run snakemake --unlock - Permission errors: Ensure write access to the working directory and
logs/
Debugging
# Dry-run to check the workflow plan
pixi run snakemake --configfile config.yaml --cores 1 -n
# Run with verbose shell commands
pixi run snakemake --configfile config.yaml --cores 8 --printshellcmds
# Generate workflow DAG
pixi run snakemake --configfile config.yaml --dag | dot -Tpdf > workflow.pdf
Citation
If you use SynFlow, please cite the relevant tools: - SyRI: Goel M. et al. Genome Biol 20, 277 (2019). doi:10.1186/s13059-019-1911-0 - MCScanX: Wang Y. et al. Nucleic Acids Res. 40(7):e49 (2012). doi:10.1093/nar/gkr1293 - DIAMOND: Buchfink B. et al. Nature Methods 18, 366–368 (2021). doi:10.1038/s41592-021-01101-x - minimap2: Li H. Bioinformatics 34(18):3094–3100 (2018). doi:10.1093/bioinformatics/bty191 - MUMmer4: Marçais G. et al. PLoS Comput Biol 14(1):e1005944 (2018).
License
This workflow is distributed under the GNU General Public License v3.0.